CE IVDR
On May 5, 2017, the EU promulgated the Regulation (EU) 2017.746 on In Vitro Diagnostic Medical Devices (IVDR), which will come into effect on May 25, 2022 and will be implemented from May 26, 2022. This means that, in May 2022, the IVDR will replace the Directive 98/79/EC of the European Parliament and of the Council on In Vitro Diagnostic Medical Devices (IVDD) promulgated by the EU in 1998, marking that the IVD management will be in the form of regulations to supervise the EU diagnostic devices.

There are many differences between the IVDR and the IVDD, for example:
· The IVDR has put forward stricter requirements for the designation of the notified bodies, and the national competent authorities and committees have strengthened the control and supervision.
· The biggest change lies in the risk classification of the in vitro diagnostic (IVD) devices and the role of the notified bodies (NBs). It also clarifies the obligations of the economic operators (manufacturers, authorized representatives, importers and distributors).
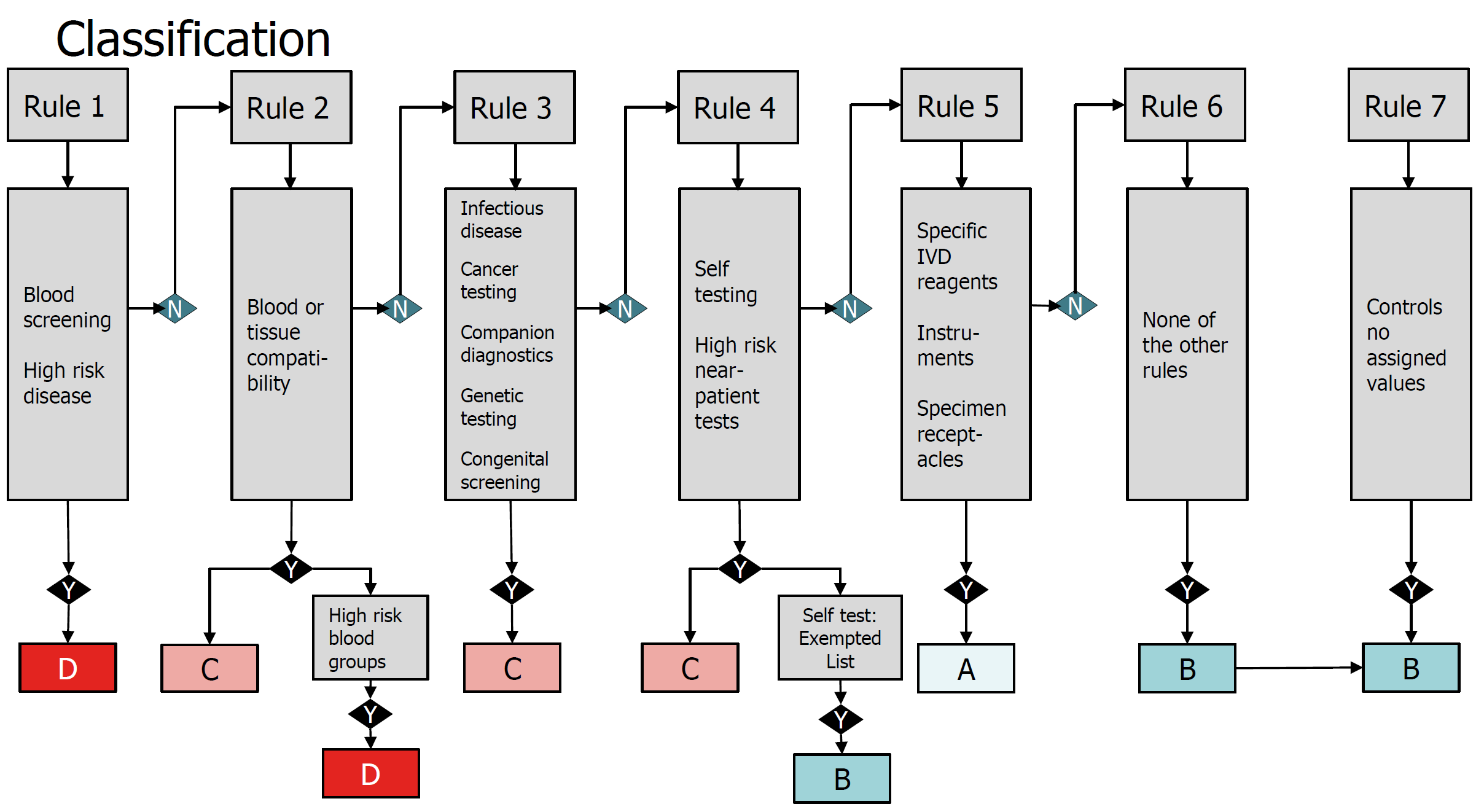
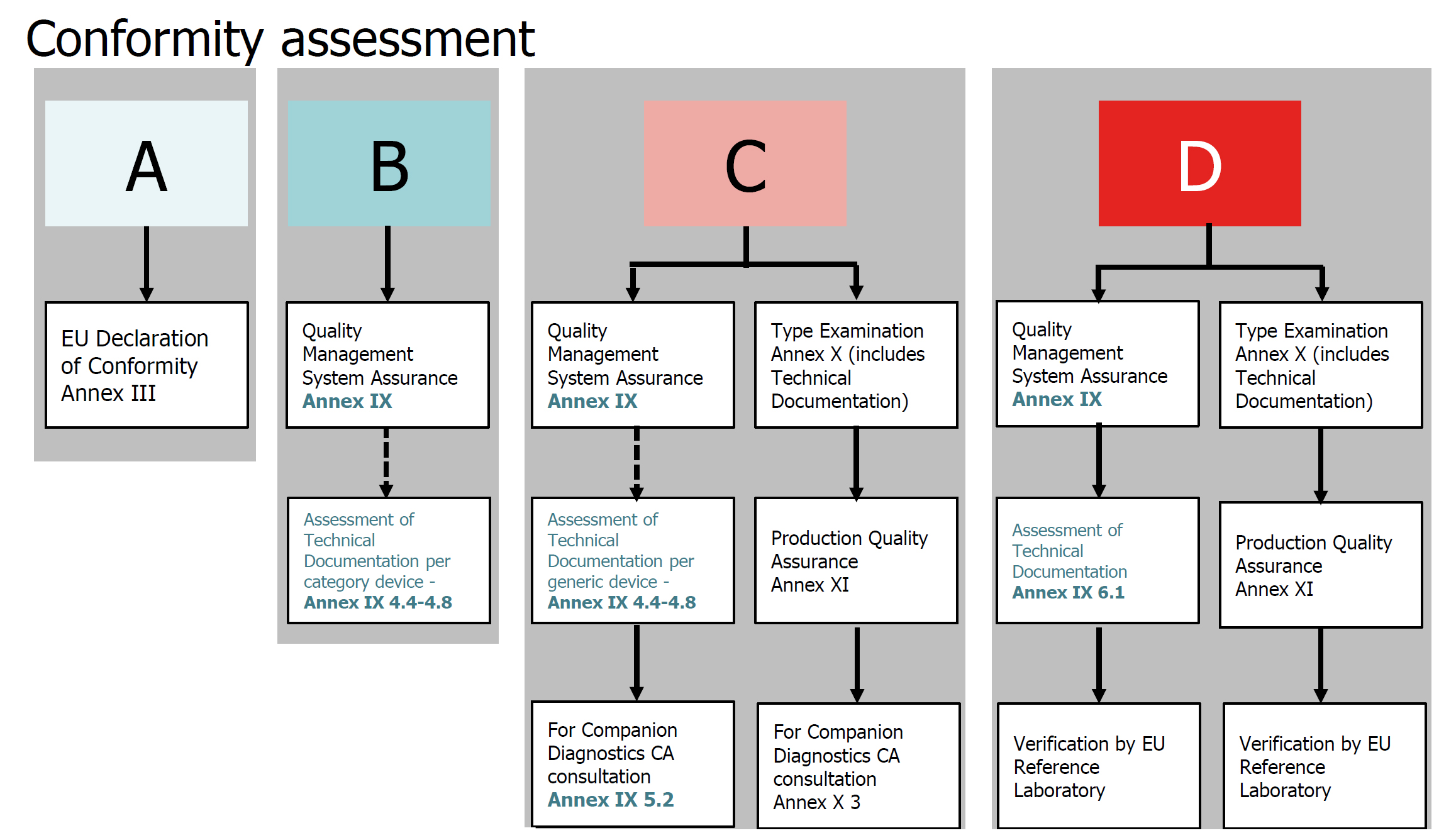
· The IVDD uses a method based on lists (List A, Lisa B and others) to categorize the risks, thereby determining the process of assessing the conformity and the degree of NB’s intervention in the supervision. The IVDR uses the internationally recognized rules to designate each device to one of the four risk categories (IVDR Article 47), ranging from A (the lowest risk) to D (the highest risk), thereby removing the vague concepts of “other categories” in the past.
· The IVDR also strengthens the requirements for the clinical evidence and conformity assessment. Approximately 85% of IVDs need supervision by the notified bodies. For companion diagnostics, the notified body shall, following the process specified in Point (k) in Section 3 of Annex X, specifically consult the competent authority designated by the member state in accordance with the Directive 2001/83/EC of the European Parliament and of the Council, or the European Medicines Agency (EMA) (Article 48).
· The conformity assessment of Class D medical devices will require the participation of an EU reference laboratory (if it is designated for this type of devices) to verify the manufacturer’s claimed performance and the conformity to the applicable general specifications (Article 48.5). In addition, currently there are no general specifications available for the innovative Class D devices, and an independent expert team is needed to provide comments on the manufacturer’s performance evaluation report (Article 48.6). Class D devices produced must be tested by an EU reference laboratory (if it is designated for this type of devices).
· The IVDR calls for greater transparency, so relevant information about IVD devices and “high-risk” performance studies will be made public. The new European Database on Medical Devices (Eudamed) will play a central role in providing more complete, accurate and easily accessible data.
· The IVDR requires the introduction of a unique device identifier (UDI) for each IVD device to significantly enhance the traceability and provide the support for post-market safety activities.
The time for the formal implementation of the IVDR in 2022 has become urgent. The brand-new product classification and conformity assessment path pose higher challenges to diagnostic medical device manufacturers. Shanghai SUNGO has an excellent team of consultants and rich case experience. We can provide you with the following consulting services.

Our consulting services
|
1 |
Professional training on the IVDR |
DAY 1 PART 1 Introduction to the IVDR PART 2 Changes in the IVDR-risk classification and conformity assessment PART 3 Changes in the IVDR-UDI and label PART 4 Changes in the IVDR-GSPR PART 5 Changes in the IVDR-PMS system PART 6 Changes in the IVDR-quality management system PART 7 Changes in the IVDR-European representative, registration, CFS |
|
DAY 2 PART 8 Changes in the IVDR-technical documentation PART 9 Changes in the IVDR-clinical evidence PART 10 Changes in the IVDR-risk management PART 11 Recommendations for the implementation of IVDR conversion PART 12 Q&A |
||
|
2 |
Special counseling |
Changes in the IVDR-UDI and label Changes in the IVDR-GSPR Changes in the IVDR-PMS and PMCF systems Changes in the IVDR-quality management system Changes in the IVDR-technical documentation Changes in the IVDR-clinical evidence and performance evaluation Changes in the IVDR-risk management |
|
3 |
European representative and registration services |
SUNGO’s companies in the Netherlands and Germany can provide the European authorized representative services and the services for registration declaration to local regulatory authorities. |
|
4 |
Services for the implementation of overall IVDR upgrade |
Covering all the contents of the above 1, 2 and 3, and also including the services for rectifying the nonconformities pointed out by the notified body after review. |
Our service process
|
1 |
Pre-evaluation |
Brief management, to ensure a clear understanding of the importance and business impact of the IVDR Organizational challenges considered in the pre-evaluation: management awareness, staffing capacity & availability, budget impact |
|
2 |
Difference analysis |
· Evaluate the impact on products, internal resources, organization and budget · Check the new classification rules (IVDR Classes A-D)
· Confirm the existing product conformity assessment path
· Check the requirements of the NB organization related to the product · Review the changes in the existing technical documents (technical documentation) · Evaluate and update the quality management system (Point 3 below) · Check the adequacy of the available clinical evidence and risk management and identify the differences (Article 56) · Evaluate the product labels (Chapter Il1, Annex I) · Ensure that the arrangements for post-market surveillance are adequate (Section 1, Chapter VII) · Develop the post-market performance follow-up (PMPF, Part B, Annex XIll) · Make provisions to meet the new vigilance requirements (Section 2, Chapter VII) · Ensure the traceability of related aspects (Chapter III) |
|
3 |
Quality system evaluation |
· Evaluate the degree of the QMS conformity to the standards and processes under the new IVDR · Add the requirements for applying the new regulations to the QMS · Assist and identify the person responsible for regulatory compliance (PPRC) and participate in training |
|
4 |
Notified body (NB) |
· Select the appropriate notified body, and confirm the qualification and scope of the notified body · Establish a transition plan for implementing the new regulations |
|
5 |
Preparation of technical documentation |
· Prepare the technical documentation (TD) that meets the requirements of the IVDR · Prepare the technical documentation such as clinical evidence and risk management · Guide the product design and development process to ensure the integrity of inputs and outputs · Confirm the labeling, post-market surveillance and post-market performance follow-up protocols · Rectify the technical documentation (risk management report, clinical performance evaluation report, GSPR, etc.) |
|
6 |
QMS |
· Update the requirements for applying the IVDR to the QMS in the existing system · Customize the corporate compliance QMS system · Implement the system implementation plan to ensure the coverage of all aspects and responsibilities |
|
7 |
Traceability UDI |
Establish the QMS requirements for traceability Establish the UDI system procedures and regulations Confirm the UDI planning and implementation |